Software

ABINIT
ABINIT is a suite of programs for materials science, which implements density functional theory, using a plane wave basis set and pseudopotentials, to compute the electronic density and derived properties of materials ranging from molecules to surfaces to solids. It implements density functional theory by solving the Kohn–Sham equations describing the electrons in a material, expanded in a plane wave basis set and using a self-consistent conjugate gradient method to determine the energy minimum. Computational efficiency is achieved through the use of fast Fourier transforms, and pseudopotentials to describe core electrons. As an alternative to standard norm-conserving pseudopotentials, the projector augmented-wave method may be used. In addition to total energy, forces and stresses are also calculated so that geometry optimizations and ab initio molecular dynamics may be carried out. Materials that can be treated by ABINIT include insulators, metals, and magnetically ordered systems including Mott-Hubbard insulators.
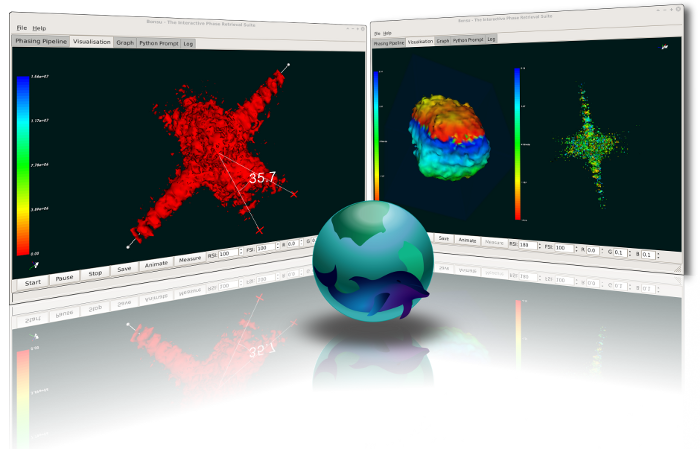
BONSU
Bonsu is an interactive phase retrieval suite, designed for phase retrieval with real-time visualisation in both two and three dimensions. It includes an inventory of algorithms and routines for data manipulation and reconstruction.

BornAgain
BornAgain is a software package to simulate and fit small-angle scattering at grazing incidence. It supports analysis of both X-ray (GISAXS) and neutron (GISANS) data. Its name, BornAgain, indicates the central role of the distorted wave Born approximation in the physical description of the scattering process. The software provides a generic framework for modeling multilayer samples with smooth or rough interfaces and with various types of embedded nanoparticles.

CONUSS
The CONUSS software package provides evaluation methods for data obtained by nuclear resonant scattering techniques. It is used for the interpretation of time or energy spectra from coherent elastic nuclear resonant scattering, i.e., forward scattering and Bragg/Laue scattering,

CrystFEL
CrystFEL is a suite of programs for processing diffraction data acquired "serially" in a "snapshot" manner, such as when using the technique of Serial Femtosecond Crystallography (SFX) with a free-electron laser source. CrystFEL comprises programs for indexing and integrating diffraction patterns, scaling and merging intensities, simulating patterns, calculating figures of merit for the data and visualising the results. Supporting scripts are provided to help at all stages, including importing data into CCP4 for further processing. [From: the Website]

Demeter
Demeter is a comprehensive system for processing and analyzing X-ray Absorption Spectroscopy data. It contains several packages such as Athena, Artemis and Hephaestus, which are widely used in the XAFS community.

easyDiffraction
easyDiffraction is scientific software for modelling and analysis of neutron diffraction data, i.e., simulation of diffraction pattern based on structural model and refinement of its parameters. easyDiffraction is similar to crystallographic programs like FullProf, Jana, GSAS, ShelX, etc. However, unlike them, easyDiffraction is based on external crystallographic libraries, such as CrysPy to cover different functionality aspects within a single intuitive and user-friendly graphical interface.

Frida
Frida (“Fast reliable interactive data analysis”) is a versatile data analysis program with special routines for inelastic neutron scattering.

MANTID
The Mantid project provides a framework that supports high-performance computing and visualisation of materials science data. Mantid has been created to manipulate and analyse neutron scattering and muon spectroscopy data, but could be applied to many other techniques.

MDANSE
MDANSE (Molecular Dynamics Analysis for Neutron Scattering Experiments) is a python application designed for computing properties that can be directly compared with neutron scattering experiments such as the coherent and incoherent intermediate scattering functions and their Fourier transforms, the elastic incoherent structure factor, the static coherent structure factor or the radial distribution function. Moreover, it can also compute quantities such as the mean-square displacement, the velocity autocorrelation function as well as its Fourier Transform (the so-called vibrational density of states) enlarging the scope of the program to a broader range of physico-chemical properties. Most of MDANSE calculations can be applied to the whole system or to arbitrary subsets that can be defined in the graphical interface while less common selections can be specified via the command-line interface. MDANSE is written in Python and currently works on Linux/debian, MacOS and Windows.

Nexus - Nuclear Elastic X-ray scattering Universal Software
The Nuclear Elastic X-ray scattering Universal Software (NEXUS) is a Python package for simulating and fitting of Moessbauer spectra, nuclear resonant scattering (NRS) data, pure electronic X-ray reflectivities (XRR), nuclear X-ray reflectivities, polarization dependent electronic scattering.
Pore3D
Pore3D is a software toolbox for quantitative analysis of three-dimensional images. The core of Pore3D consists in a set of state-of-the-art functions and procedures for performing filtering, segmentation, skeletonization and quantitative analysis of three-dimensional data. Although three-dimensional data can be produced by several techniques (for instance: magnetic resonance, x-ray scattering or confocal microscopy), the library was developed and optimized for micro-CT (Computed Tomography) data. Pore3D features are available through the high-level scripting environment IDL. Pore3D has been tested with IDL 64-bit from versions 6.4 to 8.5.

Ptychography Alignment Tools
This project provides a PyQtGraph-based GUI to assist users on the alignment of Ptychography scans. The tool has the following features: -Load set of images (tiff files supported) -Select pairs of images for alignment -Import/Export probe positions (npy array) -Image controls: levels, contrast, look up tables, zooming, translation -Preview the global picture by combining all positions

PyFAI
pyFAI is an azimuthal integration library that tries to be fast (as fast as C and even more using OpenCL and GPU). It is based on histogramming of the 2theta/Q positions of each (center of) pixel weighted by the intensity of each pixel, but parallel version uses a SparseMatrix-DenseVector multiplication

PyHST2
Hybrid distributed code for high speed tomographic reconstruction with iterative reconstruction and a priori knowledge capabilities. PyHST2 (formerly known as PyHST) has been engineered to sustain the high data flow typical of the third generation synchrotron facilities (10 terabytes per experiment) by adopting a distributed and pipelined architecture. The code implements, beside a default filtered backprojection reconstruction, iterative reconstruction techniques with a-priori knowledge. The latter are used to improve the reconstruction quality or in order to reduce the required data volume and reach a given quality goal. The implemented a-priori knowledge techniques are based on the total variation penalisation and a new recently found convex functional which is based on overlapping patches.
