Software

ABINIT
ABINIT is a suite of programs for materials science, which implements density functional theory, using a plane wave basis set and pseudopotentials, to compute the electronic density and derived properties of materials ranging from molecules to surfaces to solids. It implements density functional theory by solving the Kohn–Sham equations describing the electrons in a material, expanded in a plane wave basis set and using a self-consistent conjugate gradient method to determine the energy minimum. Computational efficiency is achieved through the use of fast Fourier transforms, and pseudopotentials to describe core electrons. As an alternative to standard norm-conserving pseudopotentials, the projector augmented-wave method may be used. In addition to total energy, forces and stresses are also calculated so that geometry optimizations and ab initio molecular dynamics may be carried out. Materials that can be treated by ABINIT include insulators, metals, and magnetically ordered systems including Mott-Hubbard insulators.

ANKAphase
ANKAphase processes X-ray inline phase-contrast radiographs by reconstructing the projected thickness of the object(s) imaged. The tool uses a single-distance non-iterative phase-retrieval algorithm described in a paper by D. Paganin et al. J. Microsc. vol. 206 (2002). It has an easy-to-use graphical user interface and can be run either as a standalone application or as a plugin to ImageJ. It works on powerful clusters but also on your office laptop.

ARCIMBOLDO
Ab Initio macromolecular phasing has been traditionally limited to small proteins at atomic resolution (1.2Å or better unless heavy atoms are present). ARCIMBOLDO constitutes a general method for 2Å data, based on combination of location of model fragments like small ?-helices with PHASER and density modification with SHELXE, distributed over a grid of computers.

Atomic Simulation Environment (ASE)
The Atomic Simulation Environment (ASE) is a set of tools and Python modules for setting up, manipulating, running, visualizing and analyzing atomistic simulations.

ATSAS
ATSAS is a program suite for small-angle scattering data analysis from biological macromolecules. It includes multiplatform data manipulation and displays tools, programs for automated data processing and calculation of overall parameters, usage of high- and low-resolution models from other structural methods, algorithms to build three-dimensional models from weakly interacting oligomeric systems and complexes, and enhanced tools to analyse data from mixtures and flexible systems.

AUTOPROC
autoPROC is a set of tools and programs to automate the whole range of steps involved in data processing: analysis of collections of images and image headers, indexing of diffraction images, determination of accurate cell parameters, integration of a series of images, processing of multi-sweep datasets, production of files of intensities and amplitudes in various formats (MTZ, Scalepack), analysis of anomalous signal, automatic determination of most likely space group symmetry.

Avizo
3D analysis software for scientific and industrial data. Different application areas: Materials Science, Quality Assurance in Industrial Environments, Electronics, Digital Rock Analysis etc. It enables users to perform interactive visualization and computation on 3D data sets. The Avizo interface is modelled on the visual programming. Users manipulate data and module components, organized in an interactive graph representation (called Pool), or in a Tree view. Data and modules can be interactively connected together, and controlled with several parameters, creating a visual processing network whose output is displayed in a 3D viewer. With this interface, complex data can be interactively explored and analyzed by applying a controlled sequence of computation and display processes resulting in a meaningful visual representation and associated derived data.

BINoculars
BINoculars is a tool for data reduction and analysis of large sets of surface diffraction data that have been acquired with a 2D X-ray detector. The intensity of each pixel of a 2D-detector is projected onto a 3-dimensional grid in reciprocal lattice coordinates using a binning algorithm. This allows for fast acquisition and processing of high-resolution datasets and results in a significant reduction of the size of the dataset. The subsequent analysis then proceeds in reciprocal space. It has evolved from the specific needs of the ID03 beamline at the ESRF, but it has a modular design and can be easily adjusted and extended to work with data from other beamlines or from other measurement techniques.
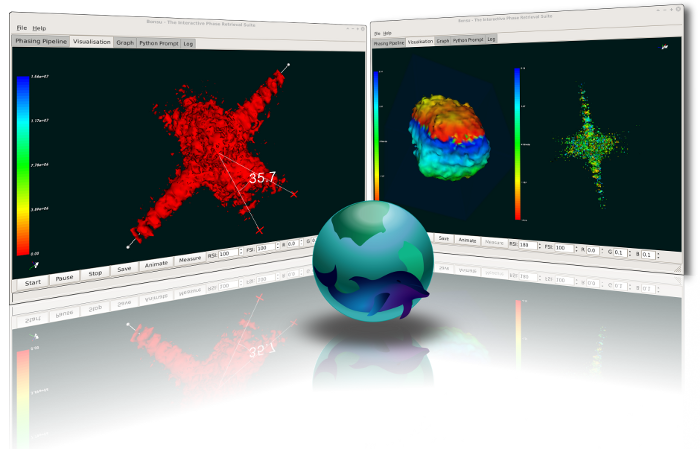
BONSU
Bonsu is an interactive phase retrieval suite, designed for phase retrieval with real-time visualisation in both two and three dimensions. It includes an inventory of algorithms and routines for data manipulation and reconstruction.

BornAgain
BornAgain is a software package to simulate and fit small-angle scattering at grazing incidence. It supports analysis of both X-ray (GISAXS) and neutron (GISANS) data. Its name, BornAgain, indicates the central role of the distorted wave Born approximation in the physical description of the scattering process. The software provides a generic framework for modeling multilayer samples with smooth or rough interfaces and with various types of embedded nanoparticles.

BUSTER
BUSTER is a framework for automatic refinement of macromolecules. It uses maximum-likelihood (ML) and maximum-entropy (ME) techniques to overcome two major shortcomings encountered by classical methods (least-squares (LS) + difference maps) when dealing with the refinement and completion of partial structures:
CCP4
CCP4 is an integrated suite of programs that allows researchers to determine macromolecular structures by X-ray crystallography, and other biophysical techniques. CCP4 aims to support the experimental determination and analysis of protein structures.

CONUSS
The CONUSS software package provides evaluation methods for data obtained by nuclear resonant scattering techniques. It is used for the interpretation of time or energy spectra from coherent elastic nuclear resonant scattering, i.e., forward scattering and Bragg/Laue scattering,

Cosmos (Lamp)
Data reduction utility to treat TOF reflectivity data. It is distributed with LAMP, but it was developed originally as a standalone IDL application and it can still be compiled and used without LAMP.

Crispy
Crispy is a modern graphical user interface to simulate core-level spectra using semi-empirical multiplet approaches.
